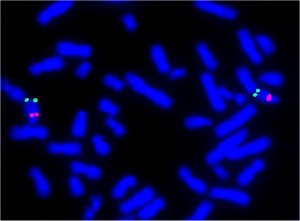
Insonnia Familiare Fatale. Sarebbe un italiano il primo uomo morto per “insonnia” E’ narrata nel libro del