
Medicina Biologica



Il tramonto delle taglie forti. Avere un seno abbondante fa male alla salute
Nonostante il diffondersi dell’ossessione per



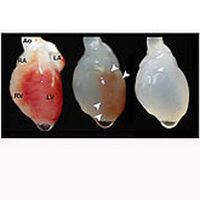
Il cuore bioartificiale: una nuova scoperta scientifica ed i possibili risvolti
L’Università del Minnesota ha condotto
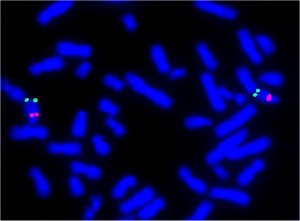



Anomalie cromosomiche. Cosa sono e quale tipo di patologie possono causare.
I cromosomi sono sono strutture
