
diagnosi prenatale
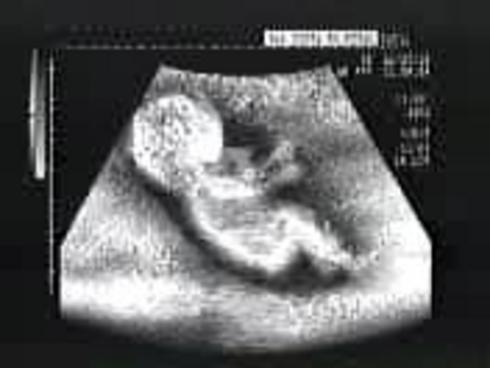
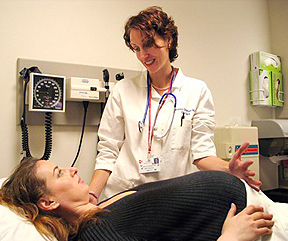
Diagnosi prenatale, fondamentale anche per donne sotto i trentacinque anni
La diagnosi prenatale, utile strumento





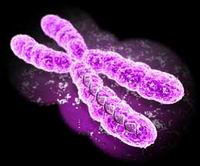
Anomalie cromosomiche. Cosa sono e quale tipo di patologie possono causare.
I cromosomi sono sono strutture

La Distrofia Muscolare di Duchenne. Come si manifesta e con quali mezzi è possibile affrontarla?
La distrofia muscolare di Duchenne