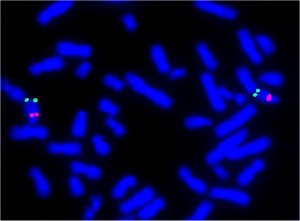
Insonnia Familiare Fatale. Sarebbe un italiano il primo uomo morto per “insonnia”
E’ narrata nel libro del

E’ narrata nel libro del
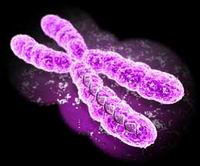
I cromosomi sono sono strutture

Gli organismi geneticamente modificati (OGM)

La distrofia muscolare di Duchenne

E’ stata diffusa recentemente la